1 Introduction to GW-BSE
1.1 Theory

where
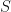 indexes the exciton state;
indexes the exciton state;  is the exciton’s center-of-mass momentum, and
is the exciton’s center-of-mass momentum, and 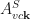 is the amplitude of the free quasi-electron and quasi-hole pair consisting of an electron in state
is the amplitude of the free quasi-electron and quasi-hole pair consisting of an electron in state  and an electron missing from state
and an electron missing from state  .
. and energy
and energy 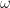 exciting an exciton from the many-body ground state. The excitonic states have an energy vs. center-of-mass momentum dispersion, and depending on the strength of the electron-hole interaction, may have multiple bound states as well as a continuum of states. The right-hand figure illustrates a exciton state as a combination of correlated interband transitions between occupied and unoccupied states in the QP bandstructure. This figure is adapted from Ref. [Cohen2016]
exciting an exciton from the many-body ground state. The excitonic states have an energy vs. center-of-mass momentum dispersion, and depending on the strength of the electron-hole interaction, may have multiple bound states as well as a continuum of states. The right-hand figure illustrates a exciton state as a combination of correlated interband transitions between occupied and unoccupied states in the QP bandstructure. This figure is adapted from Ref. [Cohen2016]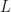 [Strinati1988]
[Strinati1988]
Here, the notation
 represents the combined time, spin, and spatial coordinate; i.e.
represents the combined time, spin, and spatial coordinate; i.e.  , and
, and  is the two-particle Green’s function. We will also use
is the two-particle Green’s function. We will also use  to refer jointly to the spin and spatial coordinate; i.e.
to refer jointly to the spin and spatial coordinate; i.e.  . The electron-hole correlation function obeys a Dyson equation known as the Bethe Salpeter equation (BSE)
. The electron-hole correlation function obeys a Dyson equation known as the Bethe Salpeter equation (BSE)
Here,
 and describes a non-interacting quasi-electron and quasi-hole pair.
and describes a non-interacting quasi-electron and quasi-hole pair. 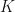 is the electron-hole interaction kernel.Following Strinati[Strinati1988] and Rohlfing and Louie[Rohlfing2000], the BSE can be written as an effective eigenvalue problem. In this form the BSE Hamiltonian has the structure
is the electron-hole interaction kernel.Following Strinati[Strinati1988] and Rohlfing and Louie[Rohlfing2000], the BSE can be written as an effective eigenvalue problem. In this form the BSE Hamiltonian has the structure
where the kernel matrix elements in each block are calculated in the basis of the single-particle orbitals. The off-diagonal blocks (
 ,
, ) can usually be neglected as long as the energy of the electron-hole interaction is small compared with the QP gap. Then, the BSE Hamiltonian becomes
) can usually be neglected as long as the energy of the electron-hole interaction is small compared with the QP gap. Then, the BSE Hamiltonian becomes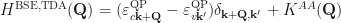
This is known as the Tamm-Dancoff approximation (TDA).The BSE kernel is found by taking the functional derivative of the self energy.
![K(3,5;4,6) = \frac{\delta[V_H(3)\delta(3,4) + \Sigma(3,4)]}{\delta G(6,5)}.](https://s0.wp.com/latex.php?latex=K%283%2C5%3B4%2C6%29+%3D+%5Cfrac%7B%5Cdelta%5BV_H%283%29%5Cdelta%283%2C4%29+%2B+%5CSigma%283%2C4%29%5D%7D%7B%5Cdelta+G%286%2C5%29%7D.+&bg=ffffff&fg=111111&s=0&c=20201002)
Within the GW approximation for $\Sigma$, the BSE Kernel becomes [Rohlfing1998,Albrecht1998,Rohlfing2000]

We refer to the first term involving the bare Coulomb interaction as the exchange kernel (
 ) and the second term involving the screened Coulomb interaction as the direct kernel (
) and the second term involving the screened Coulomb interaction as the direct kernel (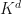 ).When the spin-orbit interaction is small, the BSE matrix can be block-diagonalized and decoupled into spin-singlet and spin-triplet classes of solution. For the singlet solutions, the BSE kernel is
).When the spin-orbit interaction is small, the BSE matrix can be block-diagonalized and decoupled into spin-singlet and spin-triplet classes of solution. For the singlet solutions, the BSE kernel is  . For the triplet solutions, there is no exchange contribution, and the BSE kernel is simply . Only the singlet states are optically bright.Once we have the solutions of the BSE Hamiltonian, we can relate them to the optical spectra. Optical absorption and conductivity are proportional to the imaginary part of the macroscopic dielectric function,
. For the triplet solutions, there is no exchange contribution, and the BSE kernel is simply . Only the singlet states are optically bright.Once we have the solutions of the BSE Hamiltonian, we can relate them to the optical spectra. Optical absorption and conductivity are proportional to the imaginary part of the macroscopic dielectric function,  . The macroscopic dielectric function is defined as
. The macroscopic dielectric function is defined as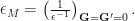
Since we are only interested in optical properties, we want to avoid having to calculate and invert
 , which is a large matrix. We use the double inversion procedure of Pick, Cohen, and Martin[Pick1970,Hanke1978,Onida2002] to directly obtain
, which is a large matrix. We use the double inversion procedure of Pick, Cohen, and Martin[Pick1970,Hanke1978,Onida2002] to directly obtain  . In this procedure, we replace the Coulomb potential in Fourier space with a modified Coulomb potential, which does not include a long-range contribution. Then, we can construct
. In this procedure, we replace the Coulomb potential in Fourier space with a modified Coulomb potential, which does not include a long-range contribution. Then, we can construct  from the solutions of the modified BSE
from the solutions of the modified BSE
where
 is the polarization vector, and
is the polarization vector, and 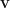 is the velocity operator. We are assuming
is the velocity operator. We are assuming 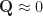 and dropping the index, since the momentum carried by light is very small.
and dropping the index, since the momentum carried by light is very small.In the independent QP picture (i.e. neglecting excitonic effects),
 is becomes
is becomes
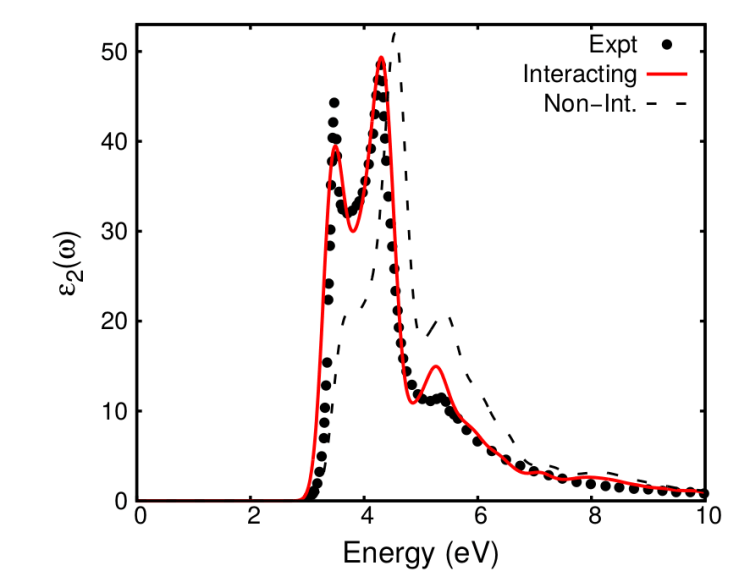
A comparison of
in the BSE and independent QP inter-band transitions picture is shown in Fig. 1. You can see that including the excitonic effects from BSE results in optical spectra in excellent agreement with experiment.
1.2 Usage in BerkeleyGW
The optical properties of materials are computed in the Bethe-Salpeter equation (BSE) executables. Here the eigenvalue equation represented by the BSE is constructed and diagonalized yielding the excitation energies and wavefunctions of the correlated electron-hole excited states. There are two main executables: kernel and absorption. In the former, the electron-hole interaction kernel is constructed on a coarse k-point grid, and in the latter the kernel is (optionally) interpolated to a fine k-point grid and diagonalized.
First, the kernel executable constructs the direct and exchange kernels as matrices in the basis of electron-hole pairs. The required input files are:
- epsmat and eps0mat: dielectric matricees from the epsilon step
- WFN_co: mean field wavefunction on a coarse k-grid
The exchange (
The kernel matrices are output in the `bsemat` file.

1.2.1 Tips for Running Kernel
- If the number of CPUs is less than the number of k-points squared (
),
and
pairs are distributed evenly over the CPUs. Thus, if you are using fewer CPUs than
, your number of CPUs should divide evenly into
, where
and
are respectively the number of valence and conduction bands.
- If each MPI task has enough memory to store the entire dielectric matrix, you should use the `low_comm` flag. This minimizes communication and makes the calculation faster.
- The kernel executable contains no check-pointing, so make sure to check your output file at the start of your calculation to see if you have enough walltime and memory to finish.
- The full list of kernel options can be found here.
The absorption code takes the bsemat file from kernel and constructs the BSE Hamiltonian. The required input files are:
- bsemat: kernel matrix
- WFN_co: the same coarse grid wavefunction used in the kernel step
- eqp_co.dat/eqp.dat (optional): QP energies from sigma on the same k-grid as WFN_co/WFN_fi
- WFN_fi (optional): wavefunction on a fine k-grid that can be used to interpolate the kernel matrix elements. This file is not needed if you choose not to interpolate (not recommended) or are studying a system without k-points.
- WFNq_fi (optional): wavefunction with a small k-shift with respect to the k-grid of WFN_fi. This is used to calculate the velocity matrix elements, which determine the oscillator strength. This file is not needed if you use choose to use the momentum operator, which neglects the nonlocal parts of the pseudopotential.
- epsmat and eps0mat: dielectric matrices from the epsilon calculation
3 References
[Albrecht1998] Stefan Albrecht, Lucia Reining, Rodolfo Del Sole, and Giovanni Onida. Ab Initio calculation of excitonic effects in the optical spectra of semiconductors. Phys. Rev. Lett., 80:4510–4513, May 1998.
[Cohen2016] M.L. Cohen and S.G. Louie. Fundamentals of Condensed Matter Physics. Cambridge University Press, 2016.
[Deslippe2012] Jack Deslippe, Georgy Samsonidze, David Strubbe, Manish Jain, Marvin L. Cohen, and Steven G. Louie. BerkeleyGW: A massively parallel computer package for the calculation of the quasiparticle and optical properties of materials and nanostructures. Comput. Phys. Commun., 183:1269, 201
[Hanke1978] W. Hanke. Dielectric theory of elementary excitations in crystals. Advances in Physics, 27(2):287–341, 1978.
[Onida2002] Giovanni Onida, Lucia Reining, and Angel Rubio. Electronic excitations: density-functional versus many-body Green’s-function approaches.
Rev. Mod. Phys., 74(2):601–659, jun 2002.
[Pick1970] Robert M. Pick, Morrel H. Cohen, and Richard M. Martin. Microscopic theory of force constants in the adiabatic approximation. Phys. Rev. B, 1:910–920, Jan 1970.
[Rohlfing1998] Michael Rohlfing and Steven G Louie. Electron-hole excitations in semiconductors and insulators. Phys. Rev. Lett., 81(11):2312–2315, 1998.
[Rohlfing2000] Michael Rohlfing and Steven G. Louie. Electron-hole excitations and optical spectra from first principles. Phys. Rev. B, 62(8):4927–4944, aug 2000.
[Strinati1988] G. Strinati. Application of the Green’s functions method to the study of the optical properties of semiconductors. Riv. Nuovo Cimento, 11:1, 1988.
