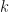Introduction to GW approximation
Theory
GW approximation is a many-body perturbation theory that describes the electron self-energy, 

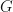


sigma_real.x (for systems with inversion symmetry) and sigma_cplx.x (for systems without inversion symmetry) are aimed to calculate the self-energy
The electron self-energy is in general non-local, and frequency-dependent. One makes several approximations (need references) and can express the electron self-energy in the following way, i.e. the so-called GW approximation,

where 
The Green’s function reads

and the screened Coulomb potential is

In DFT, people often use LDA or GGA (note: BGW also supports hybrid functional as starting point) to describe the exchange correlation in a local sense, whereas in the GW approximation, the exchange correlation is embedded in the electron self-energy. After we evaluate the 
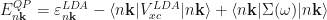
In this way, we get the quasibarticle band structures.
In BerkeleyGW, there are several different options to account for the frequency-dependence and its integral, and this is introduced in the previous section Epsilon. One can use plasmon-pole model to describe the frequency-dependence, and most often we use generalized plasmon-pole model (GPP, this is the default of BerkeleyGW); or directly sampling at different frequencies (either real or imaginary axis) and integrate, and we call this full-frequency calculation.
In GPP, one uses the dielectric matrix at zero frequency (i.e. static limit) 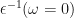
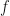


where 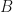
If we neglect the frequency-dependence, i.e. take the static limit, we are doing static COHSEX calculation. Furthermore, (as just touched before) if we use bare Coulomb potential, and consider only the bare exchange term (no COH, as we don’t even have 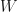
In full-frequency calculation, we separate the self-energy into bare-exchange 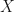

In BerkeleyGW, we calculate the physical quantities in reciprocal space with plane wave basis set.
Running tips
BerkeleyGW is designed to efficiently perform massive parallelization, so is sigma. The sigma code is parallelized over bands, and calculate the matrix elements (diagonal, or off-diagonal) one